1. Presencia de síntomas y signos de IC.
2. Función sistólica del VI normal (FE > 50 %).
3. Evidencias de anomalías de la relajación, del llenado o rigidez diastólica del VI.
La nueva clasificación pronóstica:
En fecha reciente el American College of Cardiology y la American Heart Association han enunciado una nueva clasificación de la IC basada en el desarrollo y la progresión de la enfermedad (8, 16) muy similar a las que se utilizan en el cáncer para evaluar el pronóstico desde los factores de riesgo, el carcinoma in situ asintomático hasta la diseminación metastásica.

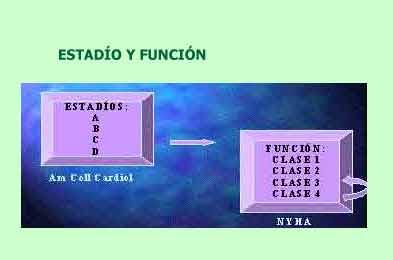
Los dos primeros estadíos (A y B) no son claramente IC pero representan la masa crítica de pacientes en riesgo y a los asintomáticos con hipertrofia del VI y/o compromiso de su función, estadíos en los que es más plausible actuar para detener la progresión y mejorar el pronóstico.
El estadío C, el de los pacientes sintomáticos, evidentemente incluye el grueso de los enfermos con IC y el estadío D, los enfermos refractarios y que actualmente se consideran tributarios de estrategias de tratamiento avanzado, y finalmente, de cuidados paliativos (16, 29).
Esta clasificación no deroga la tradicional clasificación según clases funcionales de la New York Heart Association (NYHA) sino más bien la complementa y la amplía (30). La IC puede progresar desde el estadío A hasta el D en un paciente dado, pero sin retroceso, pues del estadío D nunca podrá pasar al C, al B o al A, hágase lo que se haga. En contraste, la clasificación de la NYHA es obvio que se aplicará a los pacientes en estadíos C o D y sí puede haber mejoría de la Clase funcional con el tratamiento. Así un paciente dado, puede pasar de una Clase IV a una Clase III o II con un adecuado tratamiento.
Lo nuevo en fisiopatología:
Los dos hechos básicos que intervienen en la fisiopatología de la IC son: el remodelado ventricular y la activación de varios sistemas neurohormonales que perpetúan la disfunción, provocan la progresión de la enfermedad y son la causa de la mortalidad a largo plazo.
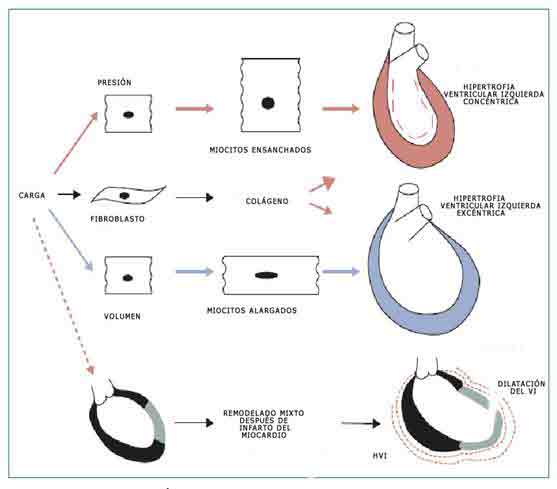
La sobrecarga de trabajo por un exceso de presión o de carga volumétrica y la pérdida de miocitos por daño isquémico (infarto) o miocardiopatía inducen el proceso de remodelado que altera el tamaño, la forma y la función ventricular.
Se reconoce que existen tres formas de remodelado: en respuesta a una sobrecarga de presión los miocitos se ensanchan produciendo una hipertrofia ventricular concéntrica, en la respuesta a la sobrecarga volumétrica ocurre un alargamiento de los miocitos con una hipertrofia excéntrica, y el remodelado que ocurre después de un infarto miocárdico que pudiéramos llamar mixto en el que alternan zonas hipertróficas con zonas dilatadas. En este proceso de remodelado también intervienen en cierta medida la fibrosis por activación de fibroblastos y la apoptosis o muerte celular programada (31).
Varios estímulos intervienen en la aparición del remodelado: unos locales, de estiramiento de los miocitos, y otros neurohormonales, como la angiotensina II, la noradrenalina, la aldosterona, las endotelinas, el óxido nítrico, el factor de necrosis tumoral a (TNF-a) y el factor de crecimiento tisular b (TGF-b) (2).
Desde el punto de vista teórico para revertir este daño inicial podrían utilizarse diversos agentes farmacológicos entre los que se incluirían los inhibidores de la enzima conversora de angiotensina (IECA), los bloqueadores adrenérgicos, los antagonistas de la aldosterona, los bloqueadores de los receptores de la endotelina, los agentes antioxidantes y los antagonistas del TNF-a. En la práctica, el tratamiento con antagonistas de la endotelina no produce beneficio clínico y puede asociarse con frecuencia a la aparición de efectos adversos, sobre todo, hepatotoxicidad (32, 33). Por otra parte, los antagonistas del TNF-a se consideran actualmente contraindicados, pues producen un aumento del riesgo de muerte u hospitalización en pacientes con IC (32, 34).
El proceso de remodelado en los pacientes con IC tiene dos fases. En la primera, los estímulos mecánicos de distensión parietal y la estimulación de los receptores neurohormonales presentes en los miocitos inducen una hipertrofia adecuada a la situación. Si ésta se mantiene crónicamente, la reserva funcional cardiaca se va agotando, los miocitos exhaustos comienzan a morir, la hipertrofia ya no es suficiente para compensar las demandas y se entra en la segunda fase, la de dilatación ventricular (31).










 Salvar en InfomeDenlaceS
Salvar en InfomeDenlaceS
 RSS Artículo
RSS Artículo